Biogen Alzheimer’s Drug Puts FDA’s Judgment in Harsh Spotlight
Biogen Alzheimer’s Drug Puts FDA’s Judgment in Harsh Spotlight
(Bloomberg) -- A new drug for Alzheimer’s disease is on a tortuous path toward the U.S. market, and while many patients would hail approval as a major advance, it could also revive concerns about regulators’ scientific integrity.
Biogen Inc. said Friday that the U.S. Food and Drug Administration is extending its review of aducanumab, an experimental therapy that patients and their families see as a potential lifeline. But the agency’s history of cherry-picking effectiveness data on the drug echoes a five-year-old decision to support a treatment for a childhood muscle-wasting disease, one that remains controversial.
FDA watchers and its own staff worry that the regulator and its leadership may be developing a pattern of approving drugs of questionable value for devastating conditions because of public pressure. Its quick authorization of Covid-19 treatments such as hydroxychloroquine that were touted by former President Donald Trump and later deemed ineffective amplifies the concern.
“No one will benefit, other than perhaps shareholders, from having a product on the market that doesn’t work,” Caleb Alexander, a Johns Hopkins University epidemiologist and FDA adviser who doesn’t think available evidence supports Biogen’s drug, said in an interview.
Both the FDA and Biogen declined to comment on their interactions.
Investor interest was piqued Friday when the agency delayed a decision by three months until June 7 on whether to clear the drug, suggesting Biogen’s application is getting additional close consideration. Biogen and partner Eisai Co. said in a statement that they gave the agency more analyses and data that will take some time to review.
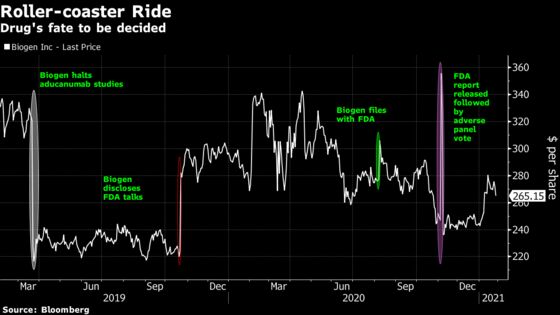
While the news met with a mixed response from analysts, Biogen shares rose 5.5% Friday. They’ve been on a roller-coaster ride since early 2019 when Biogen, based in Cambridge, Massachusetts, stopped clinical trials on aducanumab because it didn’t work in patient tests. The stock soared later the same year when a reanalysis of one of the halted trials showed a glimmer of success.
Aducanumab removes abnormal protein deposits called amyloid plaques from the brains of people with Alzheimer’s. It’s supposed to be used years before potential symptoms appear, aiming to delay patients’ decline.
Revived Hope
But debate has swirled over whether plaque removal provides any benefit. Attempts to develop similar drugs have failed, though aducanumab and potentially promising data from an experimental drug from Eli Lilly & Co. have revived some hope.
As the U.S. population ages and Alzheimer’s rates increase, an effective treatment would be a major breakthrough. The 2019 reanalysis spurred Biogen to seek FDA’s guidance, resulting in a year-long collaboration to further analyze data from the two trials, according to a document the agency and Biogen prepared ahead of a November meeting of agency advisers.
“Given the unmet medical need and unique nature of the data,” the regulator met four times with Biogen between June 2019 and June 2020 to discuss the studies, the document said. During the final meeting, they discussed the possibility of Biogen submitting an application for aducanumab approval.
Billy Dunn, the acting director of the Office of Neuroscience in FDA’s Office of New Drugs, worked with the company on the November document assessing the drug. Usually, the regulator and pharmaceutical companies write separate briefings on a drug’s data ahead of advisory panel meetings, with the FDA casting a highly critical eye.
“Our group has been testifying for five decades before FDA advisory meetings,” said Michael Carome, director of the health research group at Public Citizen, a government watchdog. “We’ve never seen such a briefing document.”
The FDA declined to make Dunn available for an interview.
When FDA’s external advisers met in November to consider recommending the drug for approval, the agency’s staff presented them with the glowing report. Evidence from the somewhat positive clinical trial was “robust and exceptionally persuasive,” the document said. It dismissed unfavorable results from a second, identically designed trial, saying they didn’t show the drug was ineffective.
Tristan Massie, an FDA statistician, dissented. “Excluding data from a large trial without sufficient justification is unscientific, statistically inappropriate and misleading,” he said in a presentation for advisory panel members.

But Massie wasn’t given much time to air his views at the November meeting, as questions were steered to the biotech’s scientists.
Agency Rebuked
“I thought the most shocking thing was when one of the panelists asked a statistical question and Dunn answered by saying, ‘You know, I’m not a statistician but maybe Biogen could answer,’” said Brian Skorney, a Robert W. Baird & Co. analyst who watched the meeting.
The panel’s discussion and recommendation against aducanumab were “a resounding and near unanimous rebuke” of the drug, Biogen and FDA’s clinical reviewers, Cowen analyst Phil Nadeau wrote at the time. But the drug could still be cleared. The agency doesn’t have to follow its advisers’ recommendations.
Biogen declined to say whether it submitted data to the FDA from another trial, called Embark, that began in March 2020. The study isn’t the most rigorous format of clinical studies because it doesn’t include a comparison group of patients who received a placebo.
About 5.8 million Americans are living with Alzheimer’s dementia, and the number is expected to balloon to almost 14 million by 2050, according to the Alzheimer’s Association. Patients and their families are searching for anything that can help stop the condition’s insidious advance.

“If you had a 50/50 chance that this drug will work for me, it’s better than the zero chance I have today,” said George Vradenburg, co-founder Us Against Alzheimer’s, a patient advocacy group.
Vradenburg has seen two relatives on his wife’s side of the family die from Alzheimer’s. Any success for aducanumab might encourage investment and further research, perhaps setting off a cascade of new treatments, he said.
Carome understands the despair; his mother died over a decade ago of Alzheimer’s after battling the disease for 10 years. Still, he’s worried about the FDA potentially approving a drug that doesn’t work.
“It could raise false hope for millions of patients and their families,” he said. “It could have adverse financial impacts and potentially bankrupt the Medicare program and, ultimately, could delay and undermine future research on Alzheimer’s drugs that could be effective.”
FDA’s handling of the situation echoes a 2016 episode when, over the objections of its own reviewers, agency leadership approved a drug for Duchenne muscular dystrophy, a lethal condition that mainly strikes young boys. Sarepta Therapeutics Inc. had only limited -- and, to FDA advisers and reviewers, unconvincing -- data on the drug’s effectiveness. But the drug, called Exondys 51, was backed by desperate parents whose sons were afflicted.
Scintilla of Effect
At the time, FDA reviewer Ellis Unger warned the decision could set a bad precedent. Future drug approvals might be based on “a mere scintilla of an effect,” he said, according to agency documents posted online.

Dunn, who was then the director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, told a panel of agency advisers convened in April 2016 that the agency’s staff had “sincere concerns” about the data behind Exondys 51. But he and Unger were overruled by FDA’s Janet Woodcock, who was just named acting commissioner by President Joe Biden.
Woodcock declined to comment. Sarepta didn’t respond when reached for comment.
Now the agency has to determine how to navigate a potential standoff on aducanumab. Massie and other critics of the drug have suggested that a third, deciding trial before an FDA decision would clear up confusion. Advocates want access for patients now. They say Biogen could conduct a study after the drug reaches the market to try to confirm aducanumab’s benefit.
Approving the drug risks sending a message that the FDA is willing to act on weak evidence, said Alexander, the Johns Hopkins-based FDA adviser.
“Clearly this is a devastating disease where new treatments are urgently needed,” he said. “For that reason it’s vital that the FDA and the marketplace gets this right.”
©2021 Bloomberg L.P.